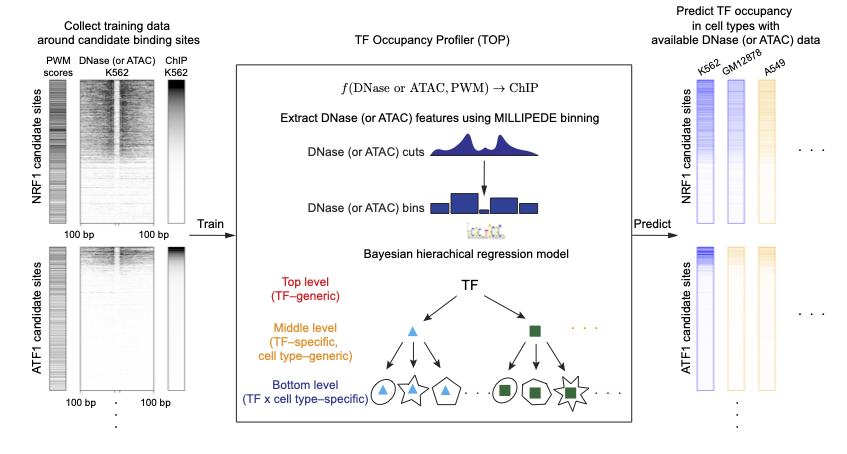
TOP fits a Bayesian hierarchical model using transcription factor (TF) motifs, DNase- or ATAC-seq data, as well as ChIP-seq data (only required in training) from multiple TFs across multiple cell types.
It can be used to predict the quantitative occupancy or binding probability for many TFs using data from a single DNase- or ATAC-seq experiment. Thus, it allows efficient profiling of quantitative TF occupancy landscapes across multiple cell types or conditions using DNase- or ATAC-seq experiments.
TOP websites
- TOP R package website
-
TOP paper resources website: a companion website for the
TOPpackage, which contains pretrained TOP model parameters and precomputed TF occupancy predictions in human, and data processing pipelines for theTOPpaper.
Install TOP R package
You can install the development version of TOP from GitHub with:
# install.packages("devtools")
devtools::install_github("HarteminkLab/TOP")After installing, check that it loads properly:
Tutorials
Please follow the tutorials to learn how to use the package.
Reference
- Luo K, Zhong J, Safi A, Hong LK, Tewari AK, Song L, Reddy TE, Ma L, Crawford GE, Hartemink AJ. Profiling the quantitative occupancy of myriad transcription factors across conditions by modeling chromatin accessibility data. Genome Res. 2022 Jun;32(6):1183-1198. doi: 10.1101/gr.272203.120.
License
All source code and software in this repository are made available under the terms of the MIT license.